

Abstract
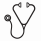
Although sickle cell disease (SCD) is a monogenic disorder, the severity and specific organ dysfunction and failure are strongly influenced by genetic modifiers. Rapid identification of all modifiers in patients and well-phenotyped cohorts will better define the impact of relevant variants on clinical status, inform disease biology, and identify new therapeutic strategies. We created the Sickle Genome Project (SGP), a whole genome sequencing (WGS) strategy, to define genomic variation and modifiers of SCD. We performed WGS on 871 African American SCD patients from St. Jude Children's Research Hospital who participated in the Sickle Cell Clinical Research and Intervention Program (SCCRIP, Hankins et al. Pediatr Blood Cancer, 2018) and Texas Children's Hospital Hematology Center (TCHC). We developed robust pipelines for accurate detection of single nucleotide polymorphisms (SNPs), identification of structural variants and data retrieval/sharing via the St. Jude Cloud platform (to be described elsewhere). Notable findings include:
1) Confirmed associations of common genetic modifiers with SCD phenotypes, including levels of fetal hemoglobin (BCL11A, HBS1L-MYB, HBB), bilirubin (UGT1A1), and microalbuminuria (APOL1). Additional associations approaching genome-wide significance require further investigation, including replication in independent samples.
2) Improved determination of the SCD modifier α-thalassemia. The most common α-thalassemia mutations in SCD are 3.7 kb or 4.2 kb deletions (-α3.7 and -α4.2 alleles), which arose from recombination between homologous HBA1 and HBA2 genes and are difficult to map using standard WGS reads. Three independent crossover events are described for -α3.7 and one for -α4.2 in SCD cohorts. We developed a novel approach to identify α-globin gene deletions by local de novo assembly of WGS data and coverage depth analysis. We identified 5 -α3.7 alleles (frequencies 0.77-32.12%) and 7 -α4.2 alleles (frequencies 0.19-5.77%). Collectively, the frequency of all -α alleles was 57%, reflecting at least 12 distinct recombination events, greatly exceeding previously published counts. These findings better define the evolution of α-globin genes to allow improved understanding of their regulation and influence on SCD.
3) Characterization of β0-thalassemia alleles. Mutations in the extended β-globin locus influence SCD phenotypes. Five SGP patients had large β-globin (HBB) deletions associated with elevated fetal hemoglobin, which ameliorates symptoms of SCD. Twenty-three patients had HbSβ0-thalassemia, which reduces the severity of some SCD phenotypes. Overall, 48.6% (18/37) of patients clinically designated as HbSβ0 -thalassemia had no identified β-thalassemia mutation. Moreover, 4/680 patients (0.6%) designated HbSS were identified to be β0-thalassemia heterozygotes. The MCV, RBC and %HbA2 distributions overlapped substantially in correct vs. incorrect genotype assignments. Improved discrimination of HbSβ0 vs HbSS genotypes by WGS will better define associated phenotype differences to impact clinical care.
4) Determination of a genetic variant linked to vaso-occlusive crisis (VOC). Previously, a single GWAS study linked rs3115229, located 63.7 kb 5′ upstream of the KIAA1109 gene, with VOC at borderline significance (P = 5.63 × 10−8) (Chaturvedi et al, Blood 130, 2017). Using WGS data for 327 SGP participants (HbSS or HbSβ0-thalassemia) enrolled in the SCCRIPP study, we found strong association (p = 7 x 10-5) between the onset of VOC and a 4-SNP diplotype within an adjacent LD block of the KIAA1109-TENR-IL2-IL21 region (chr4: 122.8Mb - 123.8Mb) which has been previously associated with numerous inflammatory disorders. We validated this association using imputed genome-wide array data in an independent group of SCD patients (Sleep and Asthma Cohort, n= 181 patients, p = 0.05) (Cohen et al, Ann Am Thorac Soc, 2016). This works provides confirmation that the region surrounding KIAA1109 is associated with pain crisis in SCD.
Our studies provide new information on the genomic architecture of SCD patients and delineate a consolidated approach for future applications of precision medicine.
Hankins:Novartis: Research Funding; Global Blood Therapeutics: Research Funding; NCQA: Consultancy; bluebird bio: Consultancy. Estepp:Global Blood Therapeutics: Consultancy, Research Funding; ASH Scholar: Research Funding; NHLBI: Research Funding; Daiichi Sankyo: Consultancy.
This icon denotes a clinically relevant abstract

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal